Mukowiscydoza (cystic fibrosis – CF) to najczęstsza wśród rasy kaukaskiej choroba uwarunkowana genetycznie. Ogromny postęp w terapii spowodował znaczny wzrost długości życia osób z mukowiscydozą, a wraz z nim zwiększenie liczby pacjentów dotkniętych powikłaniami choroby. Najczęstszym z nich jest cukrzyca (cystic fibrosis-related diabetes – CFRD). Powikłanie to istotnie wpływa na przebieg kliniczny choroby.
Praca poglądowa / Review article
Marta Fulmańska, Agnieszka Szadkowska
I Katedra Pediatrii Kliniki Pediatrii, Onkologii, Hematologii i Diabetologii
Uniwersyteckiego Szpitala Klinicznego im. Marii Konopnickiej w Łodzi
Streszczenie
Mukowiscydoza (cystic fibrosis – CF) to najczęstsza wśród rasy kaukaskiej choroba uwarunkowana genetycznie. Ogromny postęp w terapii spowodował znaczny wzrost długości życia osób z mukowiscydozą, a wraz z nim zwiększenie liczby pacjentów dotkniętych powikłaniami choroby. Najczęstszym z nich jest cukrzyca (cystic fibrosis-related diabetes – CFRD). Powikłanie to istotnie wpływa na przebieg kliniczny choroby.
Wstępne objawy CFRD są bardzo dyskretne i z tego powodu podczas diagnozowania zaburzeń tolerancji glukozy u pacjentów z CF powinien być rutynowo stosowany test doustnego obciążenia glukozą. U chorych z CFRD leczeniem z wyboru jest insulinoterapia.
Słowa kluczowe: mukowiscydoza, cukrzyca, diagnostyka, insulinoterapia
Abstract
Cystic fibrosis (CF) is the most common genetically determined disease among Caucasians. Huge progress in the treatment of this disease significantly increased the life expectancy of people affected by cystic fibrosis, thus increasing the amount of people with complications of CF. The most common complication is cystic fibrosis-related diabetes (CFRD). The initial symptoms of CFRD are very discrete and for this reason oral glucose tolerance test is routinely used for the early diagnosis of impaired glucose tolerance in patients with CF. The treatment of CFRD is the insulin therapy.
Key words: cystic fibrosis, diabetes, screening, insulin therapy
Wprowadzenie
Mukowiscydoza – zwyrodnienie torbielowate (cystic fibrosis – CF) – to najczęstsza wśród rasy kaukaskiej choroba uwarunkowana genetycznie. Dziedziczona jest autosomalnie recesywnie. Powodują ją mutacje genu CFTR, znajdującego się na chromosomie 7q31.2. Występuje z częstością 1:2500-1:4000 żywo urodzonych dzieci [1]. Ogromny postęp, jaki poczyniono w kwestii poznawania mechanizmów, a przede wszystkim terapii tej choroby, spowodował, że znacznie wzrosła średnia długość życia chorych z mukowiscydozą. Wraz z wydłużeniem długości życia pacjentów zwiększyła się jednak liczba osób dotkniętych powikłaniami CF. Najczęstszym z nich jest cukrzyca związana z mukowiscydozą (cystic fibrosis-related diabetes – CFRD) [2, 3]. Współwystępowanie cukrzycy istotnie pogarsza rokowanie u pacjentów z mukowiscydozą [4].
Częstość występowania CFRD rośnie wraz z wiekiem chorych, dotykając 40-50% dorosłych pacjentów. Cukrzyca w przebiegu CF jest często asymptomatyczna. Objawy takie jak: utrata masy ciała, uczucie zmęczenia i częste infekcje są wpisane w obraz kliniczny mukowiscydozy i mogą nie być kojarzone z cukrzycą, dopóki nie dokona się pomiaru glikemii. Poliuria i polidypsja występują stosunkowo późno. Zdarza się, że diagnoza stawiana jest dopiero po upływie 4-6 lat od pierwszych objawów [5]. Istotne było więc wprowadzenie konkretnych wytycznych dotyczących wczesnej diagnostyki i rozpoznawania CFRD.
Definicja i sposób rozpoznawania CFRD – zmiany na przełomie lat
Po raz pierwszy cukrzycę jako powikłanie mukowiscydozy opisano w 1955 roku. Mimo że cukrzycę u chorych z mukowiscydozą rozpoznaje się zgodnie z wytycznymi WHO, to ze względu na dyskretny przebieg kliniczny cukrzycy w tej grupie pacjentów oraz wpływ wczesnych zaburzeń gospodarki węglowodanowej na przebieg kliniczny choroby podstawowej dokonuje się ciągłych modyfikacji zasad prowadzenia badań przesiewowych oraz rozpoznawania CFRD. W roku 1998 problem częstości występowania, diagnostyki oraz terapii CFRD stał się przedmiotem ustaleń North American CFRD Consensus Committee. Zaproponowano wówczas podział chorych na dwie grupy: pacjentów z hiperglikemią na czczo >126 mg/dl (fasting hyperglycemia – FH+) i pacjentów bez hiperglikemii na czczo (FH-). Taka klasyfikacja została przyjęta, ponieważ podejrzewano, że prognozy u pacjentów FH+ i FH- mogą być różne.
Początkowo za badanie przesiewowe uznawano wykonywanie jeden raz do roku pomiaru przygodnej glikemii – jeśli wynosiła ona >126 mg/dl, pacjenta kwalifikowano do oznaczenia glikemii na czczo (FPG). Rozpoznanie było ustalane w momencie, gdy glikemia na czczo w dwóch pomiarach wynosiła >126 mg/dl lub gdy jednorazowy pomiar glikemii na czczo wynosił >126 mg/dl i jednorazowo stwierdzano przypadkową glikemię >200 mg/dl. Test doustnego obciążenia glukozą (OGTT) nie był polecany wówczas jako badanie przesiewowe i stosowano go tylko u osób, które nie wykazywały hiperglikemii na czczo, ale prezentowały objawy mogące świadczyć o występowaniu cukrzycy. W kolejnych latach OGTT zarekomendowano jako badanie przesiewowe dla osób z CF od 14 r.ż., a w roku 2009 według wytycznych International Society for Pediatric and Adolescent Diabetes (ISPAD) badanie to miało już obejmować dzieci od 10 r.ż. Zalecono wówczas wykonywanie pełnego testu obciążenia glukozą (dodatkowe oznaczenia glikemii w 30, 60, 90 minucie po podaniu glukozy).
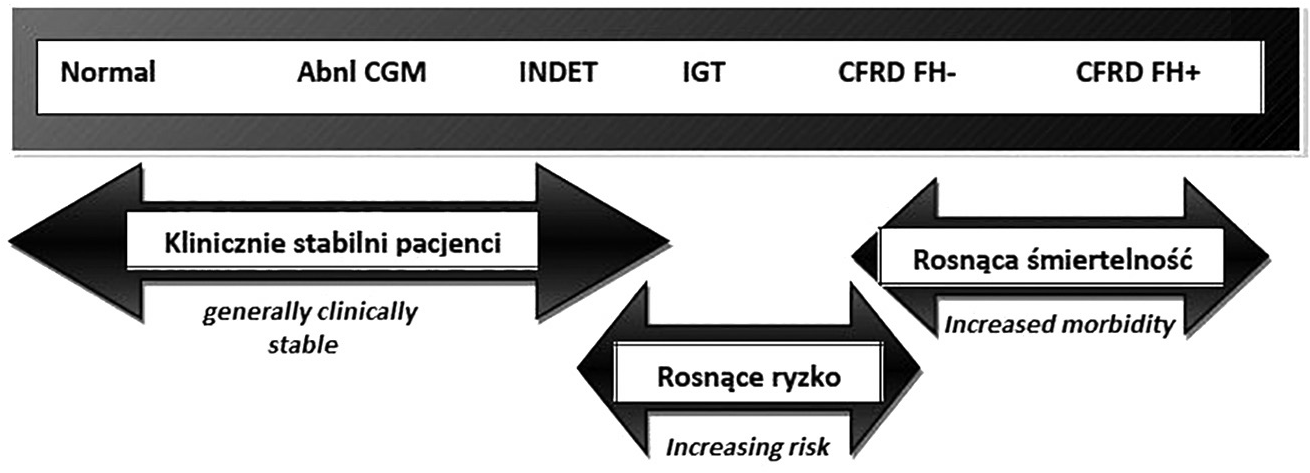
W roku 2009 podczas North American Cystic Fibrosis Consensus Conference wprowadzono kolejną jednostkę w klasyfikacji CFRD – indeterminate (INDET) (typ nieokreślony). INDET charakteryzuje się prawidłowymi wartościami glikemii na czczo oraz w 120 minucie testu OGTT przy występowaniu glikemii ≥200 mg/dl między 0 i 120 minutą testu. Rozpoznaje się go również u osób z hiperglikemią poposiłkową (≥200 mg/dl), które nie wykazują żadnych objawów klinicznych cukrzycy (ryc. 1) [6]. Na konferencji tej wskazano ponadto na zależność kryteriów rozpoznawania cukrzycy od stanu klinicznego pacjenta z CF (tab. I).
Zrezygnowano natomiast z podziału pacjentów z CFRD na FH+ i FH-. We wcześniejszych rekomendacjach pacjenci Fhnie byli kwalifikowani do terapii insulinowej. Wyniki badań udowodniły jednak korzyści płynące z wczesnego wprowadzania terapii insuliną zarówno u pacjentów FH+, jak i FH-.
Tabela I: Kryteria rozpoznawania CFRD a stan kliniczny pacjenta [6]
Table I: CFRD diagnostic criteria and clinical condition of patients [6]
|
Pacjenci leczeni ambulatoryjnie
Healthy outpatients
|
OGTT – z wyboru;
kryteria rozpoznania:
– FPG ≥126 mg/dl
– po 2 godzinach OGTT ≥200 mg/dl
– HbA1c ≥6,5%
– kliniczne objawy cukrzycy oraz przygodny pomiar glukozy ≥200 mg/dl
wszystkie oprócz ostatniego należy powtórzyć
OGTT – choice
diagnosis based on:
– FPG ≥126 mg/dL
– after 2-h OGTT ≥ 200 mg/dL
– HbA1c ≥6,5%
– random glucose ≥ 200 mg/dL plus polyuria, polydipsia
all, except the last one, should be repeated
|
|
Ciągłe żywienie pozajelitowe
Continuous drip feeding
|
diagnoza opiera się na średniej lub mierzonej
bezpośrednio po karmieniu glikemii wynoszącej
≥200 mg/dl i powinna być potwierdzona pomiarami w czasie dwóch oddzielnych nocy; jeśli
pomiary dokonywane są metodą SMBG (self-monitoring of blood glucose), należy potwierdzić je
wynikami badań laboratoryjnych
diagnosis based on mid- or immediate post-feeding: glucose ≥200 mg/dL. Should be confirmed on
two separate nights; if measured by SMBG (self-monitoring of blood glucose), should be confirmed by
laboratory measurement
|
|
Ostry przebieg choroby, stosowanie
steroidów systemowo
Acute illness, systemic steroids
|
diagnoza opiera się na stwierdzeniu hiperglikemii, która utrzymuje się przez >48 godzin;
hiperglikemia jest zdefiniowana jako:
– FPG ≥126 mg/dl
– w 120 minucie po posiłku stężenie glukozy ≥200 mg/dl; jeśli glikemia mierzona jest metodą
SMBG, należy potwierdzić uzyskane pomiary wynikami badań laboratoryjnych
diagnosis based on hyperglycemia that persists for >48 hours;
hyperglycemia is defined as:
– FPG ≥126 mg/dL
– in the 120th postprandial minute plasma glucose ≥200 mg/dL
if measured by SMBG, should be confirmed by laboratory measurement
|
|
Ciąża
Pregnancy
|
diagnoza opiera się na wykonaniu testu OGGT z 75 g glukozy; rozpoznanie ustala się w momencie
stwierdzenia któregokolwiek z następujących stężeń glukozy:
– FPG ≥92 mg/dl
– po 1 godzinie ≥180 mg/dl
– po 2 godzinach ≥153 mg/dl
diagnosis based on 75 g fasting OGGT;
if any of the following plasma glucose levels are present:
– FPG ≥92 mg/dL
– after 1 h ≥180 mg/dL
– after 2 h ≥153 mg/dL
|
Badania, takie jak pomiar HbA1c lub pomiar glikemii o dowolnej porze dnia, nie są rekomendowane w diagnostyce CFRD. Wynika to między innymi z występowania u pacjentów z CF anemii niedoborowej oraz skróconego czasu przeżycia erytrocytów, co może powodować uzyskiwanie fałszywie prawidłowych wyników pomiarów HbA1c [7]. Obecnie nie ustala się na ich podstawie rozpoznania – służą one jedynie jako badania pomocnicze. Podejrzewa się jednak, że zalecenia te mogą się zmienić. Ostatnie rezultaty badań, które przeprowadzili Burgess i wsp., wskazują na dużą wartość diagnostyczną pomiarów HbA1c oraz przygodnej glikemii jako testów przesiewowych u pacjentów dotkniętych CF. Na podstawie badań stwierdzono, że uzyskanie poziomu HbA1c ≥5,8% i przygodna glikemia >200 mg/dl z dużą dokładnością wskazują na rozpoznanie CFRD, co mogłoby zredukować potrzebę wykonywania OGTT u prawie 50% pacjentów, którzy zostali poddani badaniu [8].
Warto również wspomnieć, że zastosowanie systemu ciągłego monitorowania glikemii (continuous glucose monitoring – CGM) pozwala wykryć stan nieprawidłowej tolerancji glukozy wcześniej niż tradycyjnie wykonywany test OGTT, lecz kliniczne znaczenie zastosowania tej metody monitorowania glikemii jest wciąż badane [9-11]. Wyniki badań wskazują, że CGM jest najbardziej przydatny do wykrywania hiperglikemii, która pojawia się o określonych porach dnia, np. w godzinach wieczornych. CGM może stać się w niedługim czasie metodą z wyboru do diagnostyki zaburzeń gospodarki węglowodanowej u chorych z CF [7].
Epidemiologia
Częstość występowania CFRD różni się w zależności od wykorzystywanych badań przesiewowych i stosowanych kryteriów diagnostycznych. Choć CFRD może wystąpić w każdym wieku, częstość występowania zwiększa się z wiekiem i dotyczy 9% dzieci spośród 5-9-latków, 26% osób spośród 10-20-latków i 50% osób spośród tych, których wiek wynosi do 30 lat [12, 13]. Na podstawie okresowego powtarzania testów OGTT wykazano, że tolerancja glukozy może się zmieniać z roku na rok u pacjentów z CF [14]. W Polsce systematycznie wzrasta liczba dzieci z CFRD [15]. Część wyników badań wskazuje na nieco częstsze występowanie CFRD u dziewcząt niż u chłopców [16].
Patofizjologia CFRD
CFRD jest jednostką chorobową, która łączy w sobie cechy cukrzycy typu 1 (diabetes mellitus type 1 – DM1) oraz cukrzycy typu 2 (diabetes mellitus type 2 – DM2) (tab. II). Zaburzenia wydzielania zewnętrznego trzustki związane z nieprawidłową budową kanałów chlorkowych prowadzą do masywnego włóknienia i nacieczenia miąższu trzustki przez komórki tłuszczowe. Ten mechanizm może powodować destrukcję wysp trzustkowych [5]. Wyniki badań wykonywanych pośmiertnie dowodzą jednak, że nie ma istotnej różnicy w utracie wysp trzustkowych u osób z CFRD i u osób z prawidłową tolerancją glukozy. Uwagę zwraca natomiast występowanie białka amyloidowego, charakterystycznego dla osób z cukrzycą typu 2, aż u 69% pacjentów z CFRD i jego brak u osób bez cukrzycy [5].
Podstawowym defektem, który prowadzi do pojawienia się cukrzycy u osób z CF, jest niedobór insuliny, lecz nie obserwuje się całkowitego jej braku. Zarówno stężenia insuliny, jak i C-peptydu na czczo mogą być prawidłowe, lecz zauważa się opóźnienie szczytowego wydzielania insuliny podczas testu OGTT. Prawidłowe, szczytowe wydzielanie występuje między 30 a 60 minutą testu, natomiast u osób z CFRD odnotowuje się je między 90 a 120 minutą OGTT. To opóźnienie wiąże się z utratą pierwszej fazy wydzielania insuliny, co jest obserwowane u wszystkich pacjentów z CF, również u tych z prawidłową tolerancją glukozy [17]. Kolejną rozważaną przyczyną występowania cukrzycy u osób z CF jest insulinooporność. W wyniku badań stwierdzono zwiększoną oporność komórek wątrobowych na działanie insuliny oraz zwiększoną produkcję wątrobową glukozy [5].
Rozważano także autoimmunologiczne podłoże występowania cukrzycy w przebiegu mukowiscydozy, jednak dane zamieszczone w literaturze dotyczącej tego tematu nie są zgodne. Zwraca się dużą uwagę na występowanie i rolę, jaką odgrywają w patogenezie CFRD przeciwciała przeciwko transporterowi cynku ZnT8 (ZnT8A). Transporter ten stanowi białko transmembranowe retikulum endoplazmatycznego kodowane przez gen SLC30A8. Zmienna aktywność ZnT8 jest związana z upośledzeniem wydzielania insuliny pod wpływem glukozy. Wiązanie cynku do insuliny kontroluje jej krystalizację, proces magazynowania w komórkach ß trzustki oraz uwalnianie hormonu po posiłkach. ZnT8 został uznany za jeden z autoantygenów występujących w cukrzycy typu 1. U pacjentów z CF ZnT8A prawdopodobnie nie stanowią specyficznego indykatora pierwotnego uszkodzenia komórek ß, ale mogą obrazować stopień wtórnego uszkodzenia wysp trzustkowych, w trakcie którego dochodzi do uwolnienia autoantygenu ZnT8 [18].
Prowadzone były również badania genetyczne, których rezultaty wykazały brak związku CFRD z występowaniem genów sprzyjających pojawieniu się DM1. Odkryto jednak korelacje między CFRD a genami predysponującymi do DM2, takimi jak: gen dla TNF, heat shock protein i Calpain 10 [5]. Obecność tych genów zwiększa ryzyko i obniża średni wiek zachorowania na CFRD [7].
Stwierdzono również, że mutacja ΔF508 genu CFTR wydaje się zwiększać ryzyko wystąpienia CFRD, a mutacja N1303K może je zmniejszać. W populacjach o niskim rozpowszechnieniu ΔF508, np. w Brazylii, CFRD występuje rzadziej [7].
Powikłania CFRD i rokowanie
Do niedawna wystąpienie CFRD związane było z prawie 6-krotnie większą śmiertelnością. W 1980 roku tylko 25% pacjentów z CFRD osiągnęło 30 r.ż. w porównaniu z 60% w grupie osób bez cukrzycy. Gorsze rokowanie obserwowano głównie u kobiet. Obecnie różnica ta nie jest tak wyraźnie zaznaczona. Zmniejszyła się również różnica w przeżywalności między pacjentami z CFRD i bez cukrzycy. Wyniki badań wskazują, że wczesne rozpoznanie i agresywne leczenie odgrywają istotną rolę w poprawie przeżycia u tych chorych [19].
CFRD związana jest z gorszym odżywieniem pacjentów. Niższe wartości wskaźnika BMI obserwowane są głównie u pacjentów dotkniętych CFRD >15 r.ż. Największe zróżnicowanie w zakresie wartości tego parametru występuje wśród młodzieży między 15 a 19 r.ż. Chcąc zwrócić uwagę na rolę niedoboru insuliny w stanie odżywienia pacjentów, przeprowadzono wiele randomizowanych badań, na podstawie których stwierdzono poprawę BMI u osób dorosłych z CFRD po rozpoczęciu terapii insuliną. Prowadzi się obecnie badania w celu wykazania, że wprowadzenie do leczenia insuliny u dzieci z wczesnym jej niedoborem, jeszcze przed rozwojem CFRD, również pozytywnie wpływa na ich stan odżywienia [20].
II: Porównanie cukrzycy typu 1, cukrzycy typu 2 oraz CFRD [1]
Table II: Comparison of type 1 diabetes, type 2 diabetes and CFRD [1]
|
|
Cukrzyca typu 1
Diabetes mellitus type 1
|
Cukrzyca typu 2
Diabetes mellitus type 2
|
Cukrzyca powiązana
z mukowiscydozą
Cystic fibrosis related diabetes– CFRD
|
|
Początek / Onset
|
ostry / acute
|
powolny / insidious
|
powolny / insidious
|
|
Szczyt zachorowań /
Peak incidence
|
dzieci i młodzież
children and adolescents
|
4-6 dekada życia
4-6 decade of life
|
18-24 r.ż.
18-24 yrs
|
|
Sekrecja insuliny
Insulin secretion
|
znacznie zmniejszona lub
nieobecna
eventually absent
|
zmniejszona
decreased
|
wyraźnie zmniejszona, ale nie
nieobecna
severely decreased but not absent
|
|
Insulinowrażliwość
Insulin sensitivity
|
nieco zmniejszona
somewhat decreased
|
wyraźnie zmniejszona
severely decreased
|
nieco zmniejszona
somewhat decreased
|
|
Leczenie
Treatment
|
insulinoterapia
insulin
|
dieta, leki doustne, insulinoterapia
diet, oral meds, insulin
|
insulinoterapia
insulin
|
|
Powikłania mikroangiopatyczne
Microvascular complications
|
tak
yes
|
tak
yes
|
tak, ale w niewielkim stopniu
yes but slightly
|
|
Powikłania makroangiopatyczne
Macrovascular complications
|
tak
yes
|
tak
yes
|
nie
no
|
|
Przyczyna zgonu
Cause of death
|
choroby układu sercowonaczyniowego,
nefropatia
cardiovascular disease,
nephropathy
|
choroby układu sercowo-naczyniowego
cardiovascular disease
|
choroby układu oddechowego
pulmonary disease
|
Niezależnie od wieku chorych współwystępowanie CFRD pogarsza czynność układu oddechowego. Pogorszenie stanu klinicznego może nastąpić kilka lat przed rozpoznaniem CFRD [20]. Do niedawna objętość wydechowa pierwszosekundowa (%FEV1) była najczęściej wykorzystywanym markerem do wykrywania pulmonopatii w CF. Postępy poczynione w zakresie terapii mukowiscydozy doprowadziły do spowolnienia procesu pogarszania się funkcji płuc. Z tego powodu FEV1% stała się mniej czułym markerem progresji pulmonopatii w CF, zwłaszcza u dzieci i młodzieży. Wykonując tomografię komputerową (TK) klatki piersiowej, otrzymuje się obecnie dużo bardziej szczegółowe informacje o strukturalnych zmianach toczących się w płucach osób chorujących na CF. Zaobserwowano, że zmiany w strukturze płuc występują dużo wcześniej u osób z CFRD niż u osób z prawidłową tolerancją glukozy, podczas gdy FEV1% nie zmienia się istotnie w czasie. Wymagane są obszerniejsze dane, aby potwierdzić te wyniki i pokazać zależność czasową między rozwojem pulmonopatii a nieprawidłową tolerancją glukozy [21].
U chorych z CFRD obserwuje się również powikłania mikroangiopatyczne. Ryzyko ich wystąpienia wydaje się związane z obecnością hiperglikemii na czczo: u 16% pacjentów z CFRD FH+ stwierdzono retinopatię, a 14% miało mikroalbuminurię. U osób z CFRD FH- nie odnotowano natomiast tych powikłań. Częstość występowania mikroangiopatii jest niższa niż w przypadku innych typów cukrzycy, co może wynikać z krótszego czasu trwania cukrzycy, lepszej kontroli glikemii oraz obecności endogennego wydzielania insuliny, jak i braku innych czynników ryzyka metabolicznego, takich jak dyslipidemia i nadciśnienie tętnicze.
Neuropatia autonomiczna i gastropatia występują w CFRD równie często jak w cukrzycy typu 1. Coroczne badania przesiewowe w kierunku powikłań mikronaczyniowych są zalecane u pacjentów z CFRD, którzy mają hiperglikemię na czczo przez okres 5 lat lub od chwili rozpoznania, jeżeli dokładny moment rozpoczęcia CFRD nie jest znany [20].
Cukrzyca jest najczęstszym powikłaniem w przebiegu mukowiscydozy i zwiększa ryzyko zgonu, jednak rola hiperglikemii w przebiegu klinicznym CF pozostaje niewyjaśniona do końca [22]. Pogorszenie wydolności układu oddechowego i obniżenie wskaźników stanu odżywienia na parę lat przed rozpoznaniem cukrzycy są większe w przypadku osób z upośledzoną tolerancją glukozy niż u pacjentów z prawidłową tolerancją glukozy [23].
Terapia CFRD
Żywienie
Żywienie osób dotkniętych CFRD znacznie różni się od żywienia pacjentów z DM1 czy DM2. U chorych z CFRD nie wolno stosować ograniczeń kalorycznych [11]. Rozpoznanie i leczenie CFRD nie powinno kolidować z ogólnymi wytycznymi żywieniowymi dla osób z mukowiscydozą. Podaż kalorii u pacjentów z CF jest ściśle związana z koniecznością utrzymywania przez nich odpowiedniego BMI. Utrata masy ciała prowadzi do pogorszenia przebiegu klinicznego CF. Wskazane jest utrzymywanie BMI >50 percentyla dla płci i wieku u osób między 2 a 20 r.ż., u osób dorosłych zaś – >22 kg/m2 dla kobiet i >23 kg/m2 dla mężczyzn (tab. III).
Insulinoterapia
Terapia insulinowa jest obecnie jedyną rekomendowaną metodą leczenia pacjentów z CFRD [3]. Na podstawie przeprowadzonych badań udowodniono, że wprowadzenie terapii insulinowej u osób dorosłych dotkniętych CFRD spowodowało w ciągu 6 miesięcy znaczną poprawę funkcji układu oddechowego, przyrost masy ciała i ograniczyło katabolizm organizmu [5, 24-27].
U pacjentów z CFRD stosuje się różne modele insulinoterapii, starając się dopasować do potrzeb pacjenta. Często wykorzystywana jest metoda wielokrotnych wstrzyknięć z zastosowaniem insuliny bazalnej (insuliny NPH lub analogi insulin długodziałające) oraz doposiłkowo szybko działających analogów insulin – tzw. bolusów. Standardowa dawka insuliny bazalnej to 0,25 IU/kg m.c./24 godz. Dawka ta jest modyfikowana na podstawie pomiarów glikemii na czczo. Dawka insuliny przypadająca na bolus to 0,5-1 IU na każde 15 g spożytych węglowodanów. Dawka ta jest modyfikowana na podstawie glikemii poposiłkowych.
III: Porównanie żywienia w cukrzycy typu 1 i 2 oraz w CFRD
Table III: Comparison of nutrition in diabetes type 1 and 2 and in CFRD
|
Składniki pokarmowe
Nutriens
|
Cukrzyca typu 1 i 2
Diabetes mellitus type 1 and 2
|
Cukrzyca powiązana z mukowiscydozą
Cystic fibrosis related diabetes – CFRD
|
|
Kalorie
Calories
|
≤100% zapotrzebowania dla płci i wieku; kontrola
i ograniczanie kalorii w celu zapobiegania przyrostowi
masy ciała
≤100% of normal diet for age and gender; control or
restriction of calories to prevent the excess weight
|
najczęściej 120-150% zapotrzebowania dla płci
i wieku w celu zapobiegania utracie masy ciała
generally needs 120-150% of the basal needs for
age and gender to prevent underweight
|
|
Tłuszcze
Fat
|
<35% całkowitego zapotrzebowania energetycznego
<35% of total energy requirement
|
40% całkowitego zapotrzebowania energetycznego
40% of total energy requirement
|
|
Cukry proste
Refined sugar
|
do 10% całkowitego zapotrzebowania energetycznego
up to 10% of the total energy requirement
|
bez ograniczeń
no restriction
|
|
Cukry złożone
Carbohydrate
|
46-60% całkowitego zapotrzebowania energetycznego
46-60% of total energy requirement
|
|
Błonnik
Dietary fiber
|
brak konkretnych wytycznych; wykazano pozytywny
wpływ na przebieg choroby
no quantitative recommendation but encouraged due to
beneficial effects
|
wskazany u osób dobrze odżywionych, lecz u osób
niedożywionych może dodatkowo upośledzać
przyswajanie składników odżywczych
recommended for the well-nourished, but in the
undernourished can impair energy intake
|
|
Białko
Protein
|
10-20% całkowitego zapotrzebowania energetycznego, ale
nie więcej niż 1 g/kg m.c. w ciągu dnia
10-20% of the total energy requirement, but not over >1
g per kg of body weight per day
|
200% zalecanego spożycia
200% of reference nutrient intake
|
|
Sól / Salt
|
≤6 g w ciągu dnia / ingested ≤6 g/day
|
bez ograniczeń / without restriction
|
Zaleca się również dawki korekcyjne – 1 UI na każde 50 mg/dl powyżej 150 mg/dl glukozy we krwi. U pacjentów z CFRD dostrzega się ponadto celowość zastosowania pomp insulinowych [25, 28].
Szczególne wyzwania dotyczą prowadzenia insulinoterapii u osób, u których wymagane jest żywienie dojelitowe. U pacjentów leczonych pompami insulinowymi przy stosowaniu kilkugodzinnych wlewów dojelitowych korzystne jest zaprogramowanie bolusa złożonego, który będzie dostosowany do wartości kalorycznej pożywienia i czasu wlewu. U pacjentów leczonych metodą wielokrotnych wstrzyknięć insuliny konieczne może być podanie dawki insuliny krótko działającej.
Zaleca się, aby osoby chorujące na CFRD dokonywały pomiarów glukozy minimum trzy razy dziennie, a w czasie zaostrzeń choroby podstawowej częściej ze względu na większą skłonność do hiperglikemii w tym okresie. Uważa się, że u osób z CFRD zaostrzenie choroby podstawowej może spowodować nawet czterokrotny wzrost zapotrzebowania na insulinę.
Niewielkie zapotrzebowanie na insulinę sugeruje utrzymywanie się endogennego wydzielania insuliny zarówno u młodzieży, jak i u dorosłych z CFRD. U młodzieży resztkowe wydzielanie insuliny endogennej prawdopodobnie kompensuje insulinooporność w okresie dojrzewania, co pozwala na utrzymanie niskiego zapotrzebowania na insulinę egzogenną [29].
Leki doustne
Terapia lekami doustnymi nie jest rekomendowana w przypadku CFRD [3]. Istnieją doniesienia na temat stosowania leków doustnych w leczeniu pacjentów z CFRD, z których wynika jednoznacznie, że prowadzona za ich pomocą terapia nie była tak skuteczna i nie przynosiła tak pozytywnych efektów, jak terapia insulinowa [5]. Badaniom poddano następujące leki:
– pochodne sulfonylomocznika – nie są zalecane ze względu na duże ryzyko hipoglikemii podczas stosowania u chorych z mukowiscydozą [28],
– metformina – dolegliwości żołądkowo-jelitowe, jakie powoduje metformina, między innymi nudności, biegunki i bóle brzucha, w większości przypadków były nie do zaakceptowania przez pacjentów z mukowiscydozą [28],
– repaglinid – doustny lek hipoglikemizujący nasilający wydzielanie insuliny endogennej okazał się mniej skuteczny w porównaniu z krótko działającą insuliną w zwalczaniu hiperglikemii poposiłkowej [30],
– tiazolidinediony – nie należy ich stosować u osób z osteopenią/osteoporozą, a zatem również u osób chorujących na mukowiscydozę, w której to osteoporoza jest jednym z głównych powikłań [5].
Obecnie insulinoterapię wprowadza się po rozpoznaniu CFRD, jednak zalecenia te mogą się zmienić. U pacjentów z CF główne zaburzenia ograniczające przeżycie chorych dotyczą układu oddechowego. Prowadzone są próby określenia poziomu glikemii, przy którym dochodzi do zwiększonego ryzyka pogorszenia czynności płuc – pulmonopatii. W wydzielinie płuc jest zwykle bardzo małe stężenie glukozy, ale jeśli glikemia jest podwyższona, wydzielina może zawierać więcej glukozy, co może przyczyniać się do łatwiejszej kolonizacji układu oddechowego przez bakterie [7]. Całodobowe pomiary glikemii wskazują, że epizody hiperglikemii mogą dotyczyć jedynie okresu poposiłkowego. Wyniki prowadzonych badań dotyczących rozpoczynania insulinoterapii w stadium nietolerancji glukozy mogą wpłynąć na wybór badań przesiewowych oraz rozszerzenie wskazań terapeutycznych.
Piśmiennictwo
1. O’Riordan SM, Robinson PD, Donaghue KC, Moran A. Management of cystic fibrosis-related diabetes in children and adolescents. Pediatr Diabetes. 2009;10; suppl 12:43-50.
2. Moran A, Becker D, Casella SJ, i wsp.; CFRD Consensus Conference Committee. Epidemiology, pathophysiology, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabetes Care. 2010;33:2677-2683.
3. Moran A, Dunitz J, Nathan B, i wsp. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32:1626-1631.
4. Adler AI, Shine B, Haworth C, i wsp. Hyperglycemia and death in cystic fibrosis-related diabetes. Diabetes Care. 2011;34:1577-1578.
5. Noronha RM, Calliari LE, Damaceno N, i wsp. Update of diagnosis and monitoring of cystic fibrosis related diabetes mellitus (CFRD). Arq Bras Endocrinol Metab. 2011;55:613-621.
6. Moran A, Brunzell C, Cohen RC, i wsp.; CFRD Guidelines Committee. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010;33:2697-2708.
7. Waugh N, Royle P, Craigie I, i wsp. Screening for cystic fibrosis-related diabetes: a systematic review. Health Technol Assess. 2012;16:1-179.
8. Burgess JC, Bridges N, Gyi K, Simmonds N. Using HbAlc and random blood glucose to screen for cystic fibrosis related diabetes (CFRD). J Cystic Fibrosis. 2012;11; suppl 1:116.
9. Jefferies C, Solomon M, Perlman K, i wsp. Continuous glucose monitoring in adolescents with cystic fibrosis. J Pediatr. 2005;147:396-398.
10. Dobson L, Sheldon CD, Hattersley AT. Validation of interstitial fluid continuous glucose monitoring in cystic fibrosis. Diabetes Care. 2003;26:1940-1941.
11. O’Riordan SM, Hindmarsh P, Hill NR, i wsp. Validation of continuous glucose monitoring in children and adolescents with cystic fibrosis: a prospective cohort study. Diabetes Care.2009;32:1020-1022.
12. Moran A, Doherty L, Wang X, Thomas W. Abnormal glucose metabolism in cystic fibrosis. J Pediatr.1998;133:10-17.
13. Lanng S, Thorsteinsson B, Pociot F, i wsp. Diabetes mellitus in cystic fibrosis: genetic and immunological markers. Acta Paediatr Scand.1993;82:150-154.
14. Lanng S, Hansen A, Thorsteinsson B, i wsp. Glucose tolerance in patients with cystic fibrosis: five year prospective study. BMJ. 1995;311:655-659.
15. Fendler W, Borowiec M, Baranowska-Jaźwiecka A, i wsp. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55:2631-2635.
16. Konrad K, Thon A, Fritsch M, i wsp.; German/Austrian Diabetes Prospective Documentation Initiative. Comparison of cystic fibrosisrelated diabetes with type 1 diabetes based on a German/Austrian Pediatric Diabetes Registry. Diabetes Care. 2013;36:879-886.
17. Report of the UK Cystic Fibrosis Trust Diabetes Working Group (June 2004). http://www.fqmadrid.org/Noticias/libros%26documentos/diabetes. pdf (stan z 22.11.2013).
18. Bizzarri C, Giannone G, Benevento D, i wsp. ZnT8 antibodies in patients with cystic fibrosis: An expression of secondary beta-cell damage? J Cyst Fibros. 2013;doi: 10.1016/j.jcf.2013.03.001. [Epub ahead of print].
19. Moran A, Dunitz J, Nathan B, i wsp. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. 2009;32:1626-1631.
20. Kelly A, Moran A. Update on cystic fibrosis-related diabetes. J Cyst Fibros. 2013;12:318-331.
21. Widger J, Ranganathan S, Robinson PJ. Progression of structural lung disease on CT scans in children with cystic fibrosis related diabetes. J Cyst Fibros. 2013;12:216-221.
22. Adler AI, Shine B, Haworth C, i wsp. Hyperglycemia and death in cystic fibrosis-related diabetes. Diabetes Care. 2011;34:1577-1578.
23. Bridges N. Diabetes in cystic fibrosis. Paediatr Respir Rev. 2013;14(Suppl 1):16-18.
24. Lanng S, Thorsteinsson B, Nerup J, Koch C. Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur J Pediatr. 1992;151:684-687.
25. Nousia-Arvanitakis S, Galli-Tsinopoulou A, Karamouzis M. Insulin improves clinical status of patients with cystic-fibrosis-related diabetes mellitus. Acta Paediatr. 2001;90:515-519.
26. Rolon MA, Benali K, Munck A, i wsp. Cystic fibrosis-related diabetes mellitus: clinical impact of prediabetes and effects of insulin therapy. Acta Paediatr. 2001;90:860-867.
27. Dobson L, Hattersley AT, Tiley S, i wsp. Clinical improvement in cystic fibrosis with early insulin treatment. Arch Dis Child. 2002;87:430-431.
28. O’Riordan SM, Dattani MT, Hindmarsh PC. Cystic fibrosis-related diabetes in childhood. Horm Res Paediatr. 2010;73:15-24.
29. Sunni M, Bellin MD, Moran A. Exogenous insulin requirements do not differ between youth and adults with cystic fibrosis related diabetes. Pediatr Diabetes. 2013;14:295-298.
30. Onady GM, Stolfi A. Insulin and oral agents for managing cystic fibrosis- related diabetes. Cochrane Database Syst Rev. 2005;7:CD004730.
Adres do korespondencji:
dr hab. n. med. Agnieszka Szadkowska
I Katedra Pediatrii Kliniki Pediatrii, Onkologii, Hematologii i Diabetologii
Uniwersytetu Medycznego
ul. Sporna 36/50; 91-738 Łódź
tel.: (42) 61 77 752
e-mail: agnieszka.szadkowska@umed.lodz.pl
Konflikt interesów: nie zgłoszono
Źródło: „Przegląd Pediatryczny” 2013, vol. 43, nr 2; 82-88.